There has been a lot of concern in scientific journals and the mainstream media about colistin resistance in Enterobacteriaceae caused by plasmid-mediated resistance genes (the mcr genes). However, an article published today by our group suggests that mutational colistin resistance rather than plasmid-mediated mcr genes is a more pressing clinical threat.
We experienced an outbreak of NDM-producing K. pneumoniae CPE in 2015, which affected 40 patients mainly in renal and vascular wards over 10 months (see Figure 1). The outbreak cost the hospital >1m Euros. NDM-producing CPE tend to be resistant to a broad range of antibiotics, and most patients who required treatment (45% of the 40 in total) were treated using colistin or tigecycline.
Figure 1: Epi curve of the outbreak
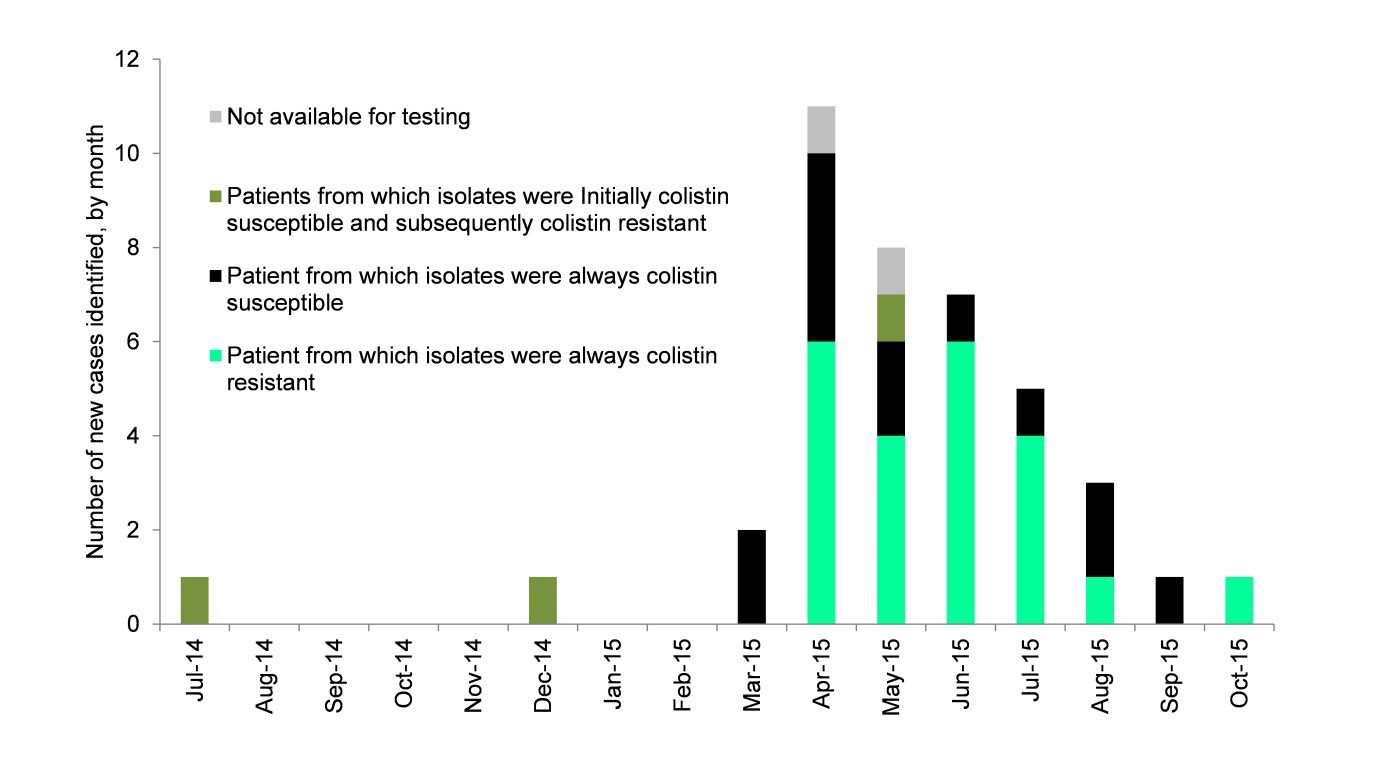
It was only late in the outbreak that we recognised that a whopping 66% of the isolates tested were resistant to colistin (25/38). Our local testing suggested that only 24% of the 38 isolates were colistin resistant. This highlights the risk of colistin resistance going undetected if laboratories are not using optimal methods for colistin susceptibility testing.
WGS analysis identified three separate potential mechanisms of colistin resistance. (‘Potential’ because whilst I am sure that these mutations do explain the colistin resistance observed in these isolates, we didn’t do the detailed molecular work to nail this association.) One of the unique aspects of the outbreak was the concurrent circulation and horizontal transmission of two closely related but distinct sub-clones on two hospital sites (see Figure 2).
Figure 2: Two overlapping, genetically related outbreaks for the price of one
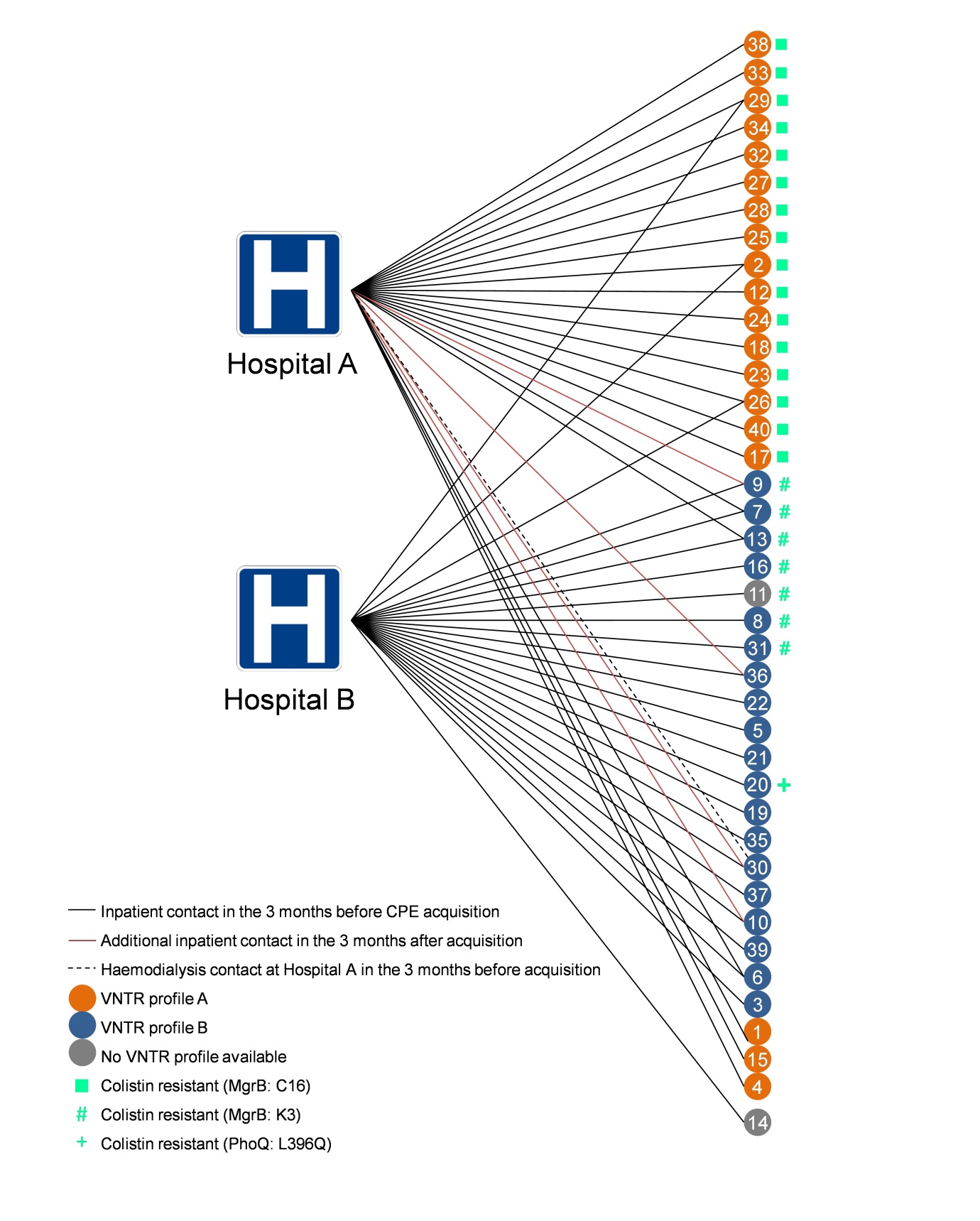
Two of the three mechanisms of colistin resistance identified appeared to spread clonally on the two separate hospital sites. Looking back, it’s quite scary to see how quickly colistin resistance emerged, and ‘watch’ the rapid clonal dissemination of the colistin-resistant sub-types across the phylogenetic tree. Incidentally, it’s never taken me so long to produce such an unsatisfactory figure (see Figure 3 below). Nobody was particularly happy with this figure throughout the review process (me included), but then nobody could think of a better way of visualising patient pathways annotated with genotypic and phenotypic (colistin-susceptibility) data. Any thoughts on how others have tackled this visualisation conundrum would be appreciated!
Figure 3: Visualising patient pathways informed with genotypic and phenotypic data.

Finally, we did not observe the high rate of mortality attributed to infection caused by CPE that has been reported elsewhere. In fact, following detailed review of the patients involved, none were considered to have died from a CPE infection. I suspect the main reason for this is that few cases were in patients on critical care units (where most CPE-related mortality has been reported), and we had no bloodstream infections.
This report should serve as a reminder that whilst plasmid-mediated resistance to colistin is clearly a problem in some parts of the world, mutational development of colisin resistance and subsequent clonal dissemination of colistin-resistant isolates is a more insidious and probably a more pressing clinical threat.
Discover more from Reflections on Infection Prevention and Control
Subscribe to get the latest posts sent to your email.
